Hombre de 60 años con erupción pruriginosa de larga evolución en las piernas: epidermólisis ampollosa distrófica
Barra lateral del artículo
Contenido principal del artículo
Resumen
Paciente masculino de 60 años, con antecedentes de hipertensión arterial, dislipidemia e hiperplasia prostática, quien tenía lesiones permanentes y muy pruriginosas en los miembros inferiores desde su juventud. Le habían tomado dos biopsias previas que fueron interpretadas como liquen plano, por lo que recibió diferentes tratamientos con corticosteroides tópicos e intralesionales, sin mejoría. Además, refirió que su padre, dos de sus hermanos y su hijo mayor tenían lesiones similares.
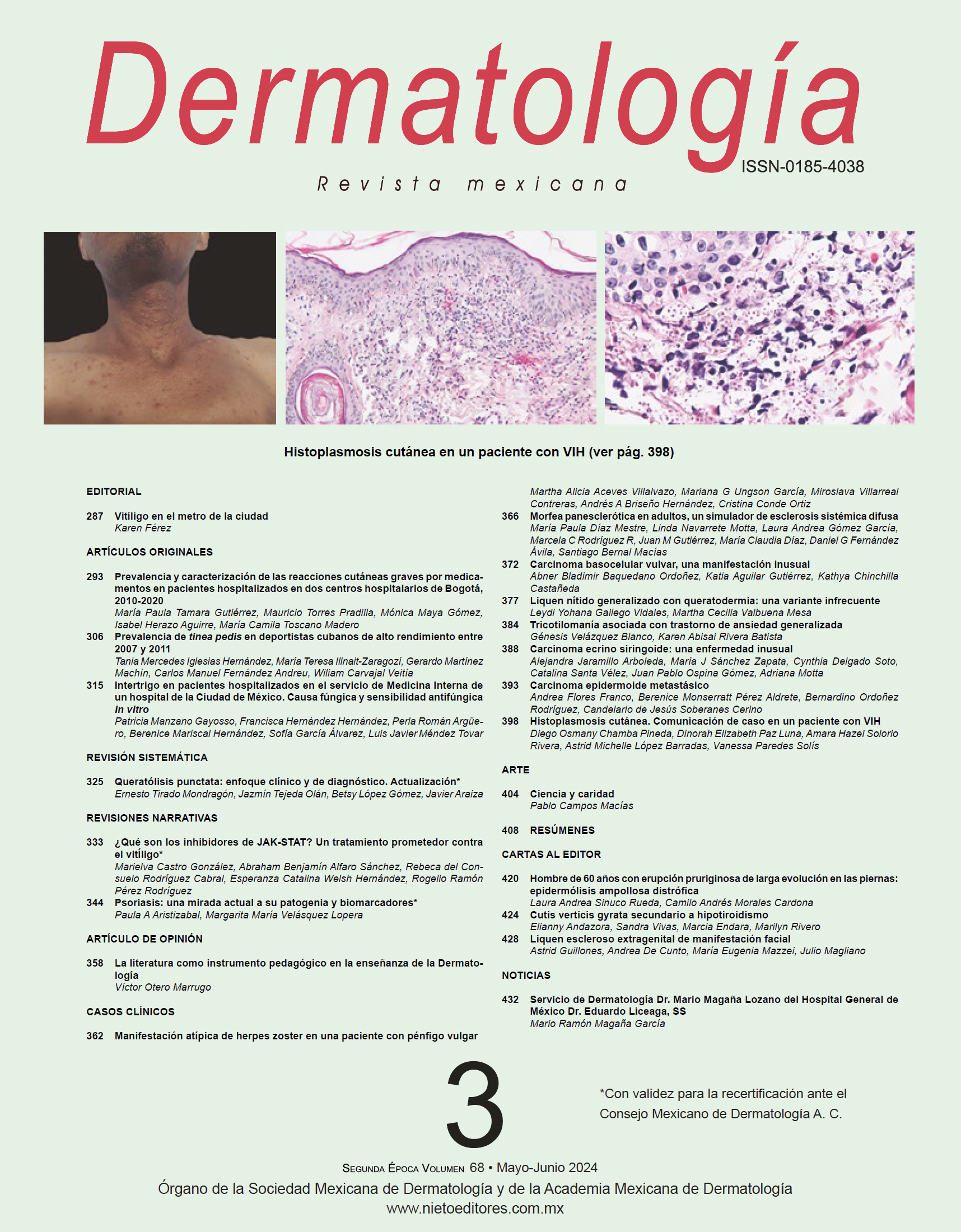
En ambos miembros inferiores tenía incontables pápulas eritematosas de color pardo, infiltradas, algunas de superficie aplanada y con descamación blanquecina, otras excoriadas y con costras hemáticas (Figura 1A y B). También se observaron cicatrices atróficas en las rodillas y en los codos, además de distrofia en algunas uñas de los pies. Con el dermatoscopio se evidenciaron escasas ampollas tensas y quistes de millium (Figura 1C). No había lesiones en las mucosas.

El estudio de la biopsia de piel reportó epidermis con formación de ampolla subepidérmica, con escasa inflamación linfocitaria y material proteináceo en su luz (Figura 2). Dermis de aspecto reparativo con neovascularización y proliferación fibroblástica. Se identificaron algunos ductos ecrinos dilatados y quistes foliculares. Las tinciones de PAS y Fite-Faraco fueron negativas para microorganismos micóticos y bacilos ácido-alcohol resistentes, respectivamente. La correlación clínico-patológica permitió confirmar el diagnóstico de epidermólisis ampollosa distrófica.

DISCUSIÓN
La epidermólisis ampollosa distrófica es una rara genodermatosis causada por una alteración en el gen que codifica para el colágeno tipo VII (COL7A1), con un espectro variable de manifestaciones clínicas.1 Esta alteración genera un déficit total o parcial del colágeno tipo VII y las fibras de anclaje, lo que se manifiesta como fragilidad cutánea y formación de ampollas subepidérmicas.2,3,4
Para clasificar la enfermedad se ha tenido en cuenta su patrón de herencia, que puede ser autosómico dominante o recesivo.2 El subtipo dominante muestra menos ampollas y se divide en la variante hiperplásica o de Cockayne-Touraine y en la albo-papuloide o de Pasini, en las que el traumatismo favorece la aparición de ampollas y cicatrices atróficas sobre las áreas extensoras.3,5 Las pápulas blanquecinas o de color marfil en el tronco y las extremidades se describieron inicialmente en la variante albo-papuloide.1
El subtipo recesivo se clasificó en severo, moderadamente severo y menos severo o, según sus características clínicas, en epidermólisis inversa, pretibial y pruriginosa, generalizada severa (Hallopeau-Siemens), generalizada intermedia, localizada, centrípeta y en dermólisis ampollosa del recién nacido.4,6,7 En la clasificación más reciente se reemplazaron los epónimos por términos más descriptivos.7 Cuadro 1

Para el diagnóstico de la epidermólisis ampollosa distrófica es fundamental la sospecha clínica y el antecedente familiar de la enfermedad, lo que favorece la correlación clínico-patológica.6 En términos clínicos se observan ampollas y cicatrices después del traumatismo, con quistes de milium, onicodistrofia o anoniquia en los pies y en algunos casos alopecia.
En las variantes epidermólisis ampollosa distrófica recesiva generalizada severa y generalizada intermedia se ha descrito anemia, retardo en el crecimiento, caries, pseudosindactilia, estenosis esofágica, malnutrición y daño ocular.4,6,7,8 Otras complicaciones asociadas son las infecciones bacterianas y mayor riesgo de carcinoma escamocelular sobre las cicatrices crónicas.8
La biopsia de piel está indicada cuando no es posible realizar el estudio genético molecular. La muestra debe tomarse del borde de una ampolla reciente (menor de 12 horas) incluyendo piel aparentemente sana.6 En la histopatología se evidencia hiperqueratosis con acantosis y la ampolla en la unión dermoepidérmica, además del infiltrado dérmico linfohistiocitario intersticial y perivascular, con fibrosis en la dermis papilar. En las lesiones albo-papuloides se observan las fibras de colágeno inmaduras y quistes de millium en la dermis superior.3
Como la microscopia de luz no permite el adecuado diagnóstico en todos los casos, la inmunofluorescencia es útil, mostrando poca o nula tinción del colágeno VII y la separación de la piel en la lámina densa.3 Los hallazgos identificados en el caso fueron los de una ampolla con desprendimiento de la unión dermoepidérmica e infiltrado linfocitario, con fibras colágenas nuevas y quistes de millium, que sugieren cambios de reparación y cronicidad. Las dos biopsias anteriores no permitieron una adecuada correlación porque fueron tomadas de lesiones papulares por sospecha de liquen plano.
No existe un tratamiento específico, pero debe prevenirse el traumatismo, controlar los síntomas y las infecciones secundarias cuando éstas ocurran.8 Para tratar el prurito se ha recurrido a diversas alternativas terapéuticas, como dapsona, colchicina, corticosteroides sistémicos, ciclosporina e inmunoglobulina intravenosa, además de rituximab en los casos más resistentes.9 La enfermedad amerita un enfoque multidisciplinario (gastroenterología, oftalmología, rehabilitación y terapia física y acompañamiento psicosocial) para evitar el deterioro en la calidad de vida de quienes la padecen.8
CONCLUSIONES
Las epidermólisis ampollosas distróficas son enfermedades poco frecuentes y de muy difícil diagnóstico cuando no se orientan a partir de la sospecha clínica. Es importante hacer una adecuada anamnesis en la que el tiempo de evolución, la historia natural de las lesiones y los antecedentes familiares proporcionan claves fundamentales. Asimismo, la elección del sitio correcto y la técnica adecuada para la toma de la biopsia favorecen la correlación clínico-patológica.